
Never Stop Learning!
|
Never Stop Learning!
|
|
In biology all of us share a basic question in biology, what properties are shared among organisms? Comparative genomics and genome sequencing allows comparison of organisms at DNA and protein levels, and sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. The sequence Comparisons can be used to: - Find evolutionary relationships between organisms - Identify functionally conserved sequences - Identify corresponding genes in human and model - organisms: develop models for human diseases Thus, sequence alignment is an important first step toward structural and functional analysis of newly determined sequences to draw functional and evolutionary inference. The sequence alignment is made between a known sequence and unknown sequence or between two unknown sequences. The known sequence is called reference sequence, and the unknown sequence is called query sequence. To proceed with the alignment process the sequences are either aligned in group of two which is called pair-wise alignment) or more than two known as, multiple sequence alignment) sequences by searching for a series of individual characters or character patterns that are in the same order in the sequences. Identical or similar characters are placed in the same column, and non-identical characters can either be placed in the same column as a mismatch or opposite a gap in the other sequence. In an optimal alignment, non-identical characters and gaps are placed to bring as many identical or similar characters as possible into vertical register. Depending upon the region of comparison, alignments are divided into two types of viz. global and local. Global Alignment Global alignment program is based on Needleman-Wunsch algorithm In global alignment, two sequences to be aligned are assumed to be generally similar over their entire length. Alignment is carried out from beginning to end of both sequences to find the best possible alignment across the entire length between the two sequences. The two sequences are treated as potentially equivalent. Goal for Global alignment: Identify conserved regions and differences, and it is applied for either comparing two genes with same function. or for comparing two sequences for conserved regions. Local Alignment Local alignment program are based on Smith-Waterman, algorithm. Local alignment does not assume that the two sequences in question have similarity over the entire length, rather, it only finds local regions with the highest level of similarity between the two sequences and aligns these regions without regard for the alignment of the rest of the sequence regions. There are three primary methods of producing local alignments, dot-matrix methods, dynamic programming, and word or k-tuple method. Goal for local alignment: The goal for local alignment is to check whether a substring in one sequence aligns well with a substring in the other, and it is applied for searching local regions of similarities in large sequences (e.g., newly sequenced genomes). or for searching conserved domains or motifs. Significance of sequence alignment Sequence alignment is useful for discovering functional, structural, and evolutionary information in biological sequences. However, it is important to obtain the best possible or “optimal” alignment to discover this information. Sequences that are very much alike, or “similar” in the parlance of sequence analysis, probably have the same function or there may have been a common ancestor sequence, and thus, the sequences are then defined as being homologous. The alignment indicates the changes that could have occurred between the two homologous sequences during the course of evolution. Now let us learn more about sequence alignment using this video tutorial.
0 Comments
BLAST (basic local alignment search tool) is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA or RNA sequences. BLAST performs “local” alignments, and this is particularly helpful when working with one or more functional domains occurring within a protein. The BLAST algorithm is tuned to find these domains or shorter stretches of sequence similarity. Moreover, the local alignment approach also means that an mRNA can be aligned with a piece of genomic DNA, as its is frequently required in genome assembly and analysis. BLAST works by finding regions of local similarity between sequences comparing nucleotide or protein sequences to sequence databases and it also calculates the statistical significance of matches, and displays a “expect value” or e-value that estimates how many matches would have occurred at a given score by chance, which can aid a user in judging how much confidence to have in an alignment. Uses of BLAST BLAST can be used for several purposes such as identification of species, locating domains, establishing phylogeny, DNA mapping, and Sequence comparisons. Identification of species: With the use of BLAST, we can correctly identify a species or find homologous species. This can be useful, for example, when you are working with a DNA sequence from an unknown species. Locating domains: When working with a protein sequence you can input it into BLAST, to locate known domains within the sequence of interest. Establishing phylogeny: Using the results received through BLAST you can create a phylogenetic tree using the BLAST web-page. Phylogenies based on BLAST alone are less reliable than other purpose-built computational phylogenetic methods, so should only be relied upon for "first pass" phylogenetic analyses. DNA mapping: When working with a known species, and looking to sequence a gene at an unknown location, BLAST can compare the chromosomal position of the sequence of interest, to relevant sequences in the database(s). NCBI has a "Magic-BLAST" tool built around BLAST for this purpose.[30] Sequence Comparison: When working with genes, BLAST can locate common genes in two related species, and can be used to map annotations from one organism to another. Therefore BLAST has proven itself to be an important tool for studying functional and evolutionary relationships between sequences as well as help identify members of gene families. 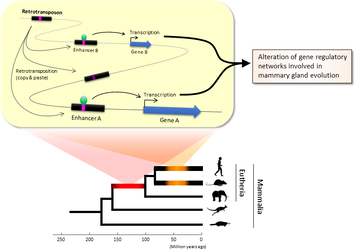 Figure 1. Retrotransposons have been co-opted to act as enhancers in the mammalian genome. Transposable elements known as retrotransposons duplicate themselves via a copy-and-paste mechanism. Tens of thousands of these sequences were found to contain binding sites for proteins that act as master regulators of mammary gland development. Figure 1. Retrotransposons have been co-opted to act as enhancers in the mammalian genome. Transposable elements known as retrotransposons duplicate themselves via a copy-and-paste mechanism. Tens of thousands of these sequences were found to contain binding sites for proteins that act as master regulators of mammary gland development. Transposable elements (TEs), so-called selfish DNA sequences, are known to be capable of moving around the genome through cut-and-paste or copy-and-paste mechanisms, and our human genome contains approx 4.5 million copies of these TEs. This can not be termed as one obscure event as they account for 30-50% of mammalian DNA. The trnasposable elements have been traditionally considered as genetic freeloaders hitchhiking along in the genome without providing any benefit to the host organism. More recently, however, scientists have begun to uncover cases in which TE sequences have been co-opted by the host to provide a useful function, such as encoding part of a host protein. In a recent study published in the journal Nucleic Acids Research, Professor Hidenori Nishihara from Department of Life Science and Technology, Tokyo Institute of Technology, who has undertaken one of the most comprehensive analyses of TE sequence co-option to date, uncovers tens of thousands of potentially co-opted TE sequences and the findings suggest that the TEs might have played a key role in mammalian evolution. Talking about his research Professor Nishihara says that "I was specifically interested in the potential influence of TE sequences on the evolution of the mammary gland, an organ that is responsible for producing milk and is, as the name suggests, a key distinguishing feature of mammals." To identify potentially co-opted TE sequences, Dr. Nishihara used four proteins—ERα, FoxA1, GATA3, and AP2γ—that bind to DNA to regulate the production of proteins involved in mammary gland development, and located all of the DNA sequences in the genome to which these proteins bind. Surprisingly, 20–30% of all of the binding sites across the genome were located in TEs, with as many as 38,500 TEs containing at least one binding site. The majority of these were in a copy-and-paste type of TE known as a retrotransposon, which duplicates itself, leaving a new copy in a new location. The TE-derived binding site sequences were more conserved across species than expected, indicating that they are being preserved by evolution because they serve some important function. Dr. Nishihara believes that these TE sequences have been co-opted to serve as enhancers, DNA elements that increase the transcription of nearby genes (Fig. 1). By binding to one of the four master regulators of mammary gland development, these enhancers ultimately increase the production of proteins involved in mammary gland development. Dr. Nishihara then investigated when in mammalian evolution these TE sequences were acquired and found two distinct phases of acquisition: roughly 60–70% were acquired in the ancestor of all placental mammals (Eutheria), while 10–20% could be traced back to the ancestor of New World monkeys (Simiiformes) (Fig. 2, left). In addition, there appeared to be another wave of acquisition of ERα binding sites in the ancestor of mice and rats (Muridae) (Fig. 2, right). Thus, by providing a vast number of potential regulatory element binding sites throughout the genome, TEs may have had a substantial impact on the emergence of the mammary gland and its evolution within mammals. 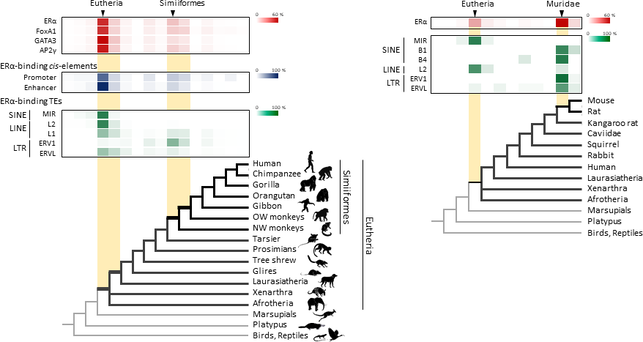 Figure 2. Transposable element-derived binding sites were acquired during distinct phases in mammalian evolution. Left: Among the TE-derived binding sites identified, 60–70% were acquired in the ancestor of placental mammals (Eutheria), while 10–20% were acquired in the ancestor of New World monkeys (Simiiformes). Right: Many ERα binding sites were also acquired in the ancestor of mice and rats (Muridae). Dr. Nishihara's study sheds light on the deep involvement of TEs in the evolution of mammary gland regulatory elements. However, it remains unclear how common this mode of TE-mediated regulatory network evolution is. Dr. Nishihara, at least, believes that the mammary gland is not unique in this respect. He notes that, "in addition to mammary glands, mammals share many features, such as the neocortex, closed secondary palate, and hair. I expect future research to uncover many additional kinds of TEs that have been similarly involved in the evolution of these features in mammals." References

The beginning of a long quest
It was the year 1856 when few limestone excavators working near Düsseldorf, Germany, unveiled bones that resembled to humans and initial analysts inferred them as belonging to a deformed human, citing their oval shaped skull, with a low, receding forehead, distinct brow ridges, and bones that were unusually thick. It was only subsequent studies that revealed that the remains belonged to a previously unknown species of hominid, or early human ancestor, that was similar to our own species, Homo sapiens. In 1864, the specimen was dubbed Homo neanderthalensis, after the Neander Valley where the remains were found.Neanderthals rose to prominance around 200,000 and 250,000 years ago and ruled the hills and grasslands of europe till extiction around 30000 years ago. The exact date of their extinction had been disputed but in 2014, a team led by Thomas Higham of the University of Oxford used an improved radiocarbon dating technique on material from 40 archaeological sites to show that Neanderthals died out in Europe between 41,000 and 39,000 years ago, with the last group disappearing from southern Spain 28,000 years ago. Similarity of Neanderthals with Rhodesian Man (Homo rhodesiensis) made early investigators infer that they share similar ancestor but comparison of the DNA of Neanderthals and Homo sapiens suggests that they diverged from a common ancestor between 350,000 and 400,000 years ago, which some argue might be Homo rhodesiensis but this argument assumes that H. rhodesiensis goes back to around 600,000 years ago. However one can not rule out convergent evolutionary paths for the two hominids displaying feathres such as distinct brow ridges. Neanderthals settled in Eurasia, but not extending beyond modern day Israel. No neanderthal sites were observed in the African continent and Homo sapiens appears to have been the only human type in the Nile River Valley because of the warmer climate present in that period. Are Neanderthals really extinct? Sudden disappearnce of Neanderthals from Europe co-incides with the arrival of H. sapiens and this information prompted many scientists to suspect that the two events are closely linked, and humans contributed to the demise of their close cousins, either by outcompeting them for resources or through open conflict. The hypothesis that early humans violently replaced Neanderthals was first proposed by French palaeontologist Marcellin Boule (the first person to publish an analysis of a Neanderthal) in 1912. However according to a 2014 study by Thomas Higham and colleagues based on organic samples suggest that the two different human populations shared Europe for several thousand years. Therefore outright violent extinction seems less plausible and leads to the formation of two scenarios for Neanderthal extinction. Possible scenarios for the extinction of the Neanderthals are:
Ancient DNA to the rescue
DNA sequence analysis of the fossils can reveal an entirely new world of information to us, but recovering DNA from samples that are fossilized thousands of years ago, is a daunting task in itself making ancient DNA research far from routine. The samples are prone to degradtion and contamination by DNA from other sources, and retriving data out of the ancient material is costly and painstaking work. At a more fundamental level, it requires determining whether the necessary samples even exist and, if so, how to get access to them. The revelations An international group of Anthropologists from Max Planck Institute for Evolutionary Anthropology, Cold Spring Harbour Laboratories and Cornell University using various different methods of DNA analysis estimated an interbreeding to have happened less than 65,000 years ago, around the time that modern human populations spread across Eurasia from Africa. They reported evidences for a modern human contribution to the Neanderthal genome. Martin Kuhlwilm, co-first author of the new paper, identified the regions of the Altai Neanderthal genome sharing mutations with modern humans. They found evidences of gene flow from descendants of modern humans into the Neanderthal genome to one specific sample of Neanderthal DNA recovered from a cave in the Altai Mountains in southern Siberia, near the Russia-Mongolia border. Earlier studies have observed that DNA of modern humans contains 2.5 to 4 percent Neanderthal DNA. However studies conducted by Mendez et. al. revealed that no Neanderthal Y chromosomal DNA was ever observed in any human sample they have tested. Contemplating upon the observations they initially felt that the Neanderthal Y chromosome genes could have drifted out of the human gene pool by chance over the millennia, or there are possibilities that the Neanderthal Y chromosomes include genes that are incompatible with other human genes. Mendez, and his colleagues have found evidence supporting this idea, and they think that the two groups may have been reproductively isolated unlike thought earlier. Their study identified protein-coding differences between Neandertal and modern human Y chromosomes. Changes included potentially damaging mutations to PCDH11Y, TMSB4Y, USP9Y, and KDM5D, and three of these changes are missense mutations in genes producing male-specific minor histocompatibility (H-Y) antigens. Antigens derived from KDM5D, for example, are thought to elicit a maternal immune response during gestation. It is possible that these incompatibilities at one or more of these genes played a role in the reproductive isolation of the two groups. Thus Y-chromosomal studies have re-drawn the time-line of divergence of the two species ~4 million years ago, which according to previous estimates based on mitochondrial DNA put the divergence of the human and Neanderthal lineages at between 400,000 and 800,000 years ago. New data emerging out of GWA studies could shed further light on the evolutionary history of the two hominids. In my opinion the image could resolve better if we look into the pathogen associated and immune response genes that we might have inherited or acquired during our evolutionary journey. Reference:
|
AuthorHello! My name is Arunabha Banerjee, and I am the mind behind Biologiks. Leaning new things and teaching biology are my hobbies and passion, it is a continuous journey, and I welcome you all to join with me Archives
June 2024
Categories
All



|
 RSS Feed
RSS Feed
