
Never Stop Learning!
|
Never Stop Learning!
|
|
Most of us grow up listening, reading and learning the fact that we "Human" beings can boast of being the most evolved and a higher organism. Earlier than 1960, the image below would have been correct and sensible. However our idea of supremacy in terms of genome size and number of genes takes a flak when we first started looking at the complexity of genome size, it was soon realized that the large genomes were often composed of huge chunks of repetitive DNA, while only a few percent of the genome in these organisms were unique. Now lets have a look at the past and try to figure out the origin of this concept. 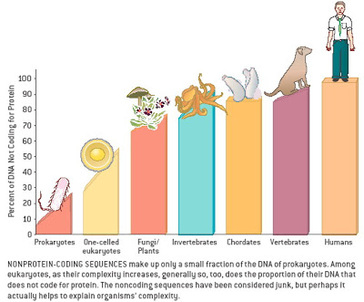 Classically biologists recognize that the living world comprises two types of organisms. Prokaryotes and Eukaryotes. Assuming that you already know what prokaryotes and eukaryotes are, I am not going to dive into the difference between the two. So what is C-value? 'C-value ', of an organism is defined as the total amount of DNA contained within its haploid chromosome set. Prokaryotic cells typically have genomes smaller than 10 megabases (Mb), while the genome of single cell eukaryote is typically less than 50Mb. Therefore for simplicity's sake here we are not comparing the genomes from two classes of organisms together. However eukaryotes alone show immense diversity among their genome sizes, from the smallest eukaryote being less than 10 Mb in length, and the largest over 100 000 Mb, and all these observation seems coinciding to a certain extent with the complexity of the organism, the simplest eukaryotes such as fungi having the smallest genomes, and higher eukaryotes such as vertebrates and flowering plants having the largest ones. So it seems fair to think that, complexity of an organism is related to the number of genes in its genome - higher eukaryotes need larger genomes to accommodate the extra genes. However, in fact this correlation is far from being precise: if it were, then the nuclear genome of the yeast S. cerevisiae, which at 12 Mb is 0.004 times the size of the human nuclear genome, would be expected to contain 0.004 × 35 000 genes, which is just 140. In fact the S. cerevisiae genome contains about 5800 genes! Therefore for many years this lack of precise correlation between the complexity of an organism and the size of its genome was looked on as a bit of a puzzle, and called as C-value paradox/C-value enigma. Questions raised by C-value paradox The C-value paradox not only represents one question, but it rather raises three of them, as suggested by T. R. Gregory(2007), 1) the generation of large-scale variation in genome size, which may occur by continuous or quantum processes, (2) the non-random distributions of genome size variation, whereby some groups vary greatly and others appear constrained, and (3) the strong positive relationship between C-value and nuclear and cell sizes and the negative correlation with cell division rates. Therefore any proposed solution must try to solve these three problems as well. Now we biologists are pretty good at dividing ourselves among different school of thoughts (remember the RNA and DNA world!) and the C-value paradox wasn't an exception either. Nonetheless two school of thoughts emerged here too, one proposing Mutation pressure theories and the other proposing Optimal DNA theories. The table shown below summarizes the theories proposed along with their proposed mechanism. Each theory can be classified according to its explanation for the accumulation or maintenance of DNA (MP,mutation pressure theory ; OD, optimal DNA theory) and according to its explanation for the observed cellular correlations (CN, coincidental, CE, coevolutionary, CA, causative). Note that these theories are not necessarily mutually exclusive in all respects, since the optimal DNA theories do not specify the mechanism(s) of DNA content change and can include those presented by both mutation pressure theories. (ref: GREGORY, T. R. (2001),page # 69) 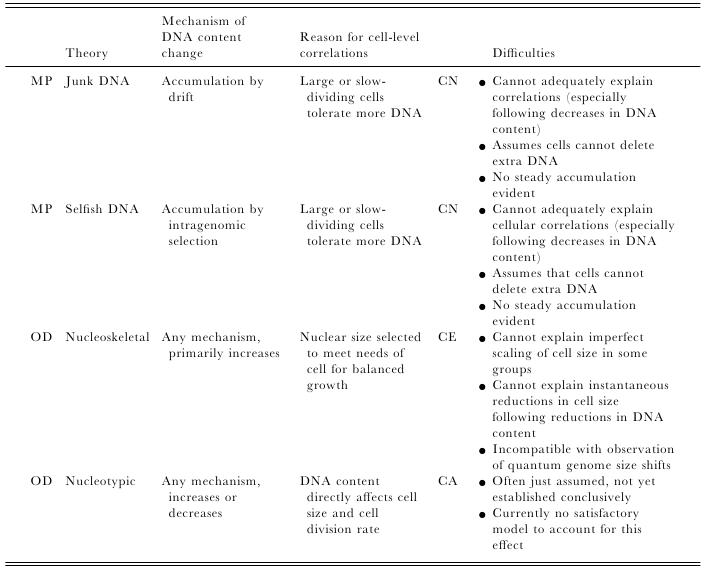 So what is the most plausible explanation of C-value paradox? In 1980s, two landmark papers, by Orgel and Crick and by Doolittle and Sapienza, established a strong case against 'selfish DNA elements' which we better know as transposons. They proposed that ‘selfish DNA’ elements, such as transposons, essentially act as molecular parasites, replicating and increasing their numbers at the (usually slight) expense of a host genome, i.e these elements functions for themselves while providing little or no selective advantage to the host. Computational genomic studies have shown that transposable elements invade in waves over evolutionary time, sweeping into a genome in large numbers, then dying and decaying away leaving the 'Junk DNA' in its trail. 45% of the human genome is detectably derived from these transposable elements. Therefore we can say that C-value paradox is mostly (though not entirely) explained by different loads of leftovers from transposable elements and larger the genomes longer is the trail leftover by transposons. So, if the C-value paradox is explained and rested for good, why dig it up again? Recent publications discussing the outcome of ENCODE (Encyclopedia Of DNA Elements) project suggest that 80% of the human genome is reproducibly transcribed, bound to proteins, or has its chromatin specifically modified. Moreover the previously considered junk DNA is found to be biochemically active disapproving the 'Junk DNA' theory. Now in the light of ENCODE data it is pertinent that scientists need to come up with a alternative hypothesis capable of explaining C-value paradox, for mutational load, and for how a large fraction of eukaryotic genomes is composed of neutrally drifting transposon-derived sequences. Reference:
1 Comment
Sankar
8/6/2018 12:16:09 am
Super explanation Leave a Reply. |
AuthorHello! My name is Arunabha Banerjee, and I am the mind behind Biologiks. Leaning new things and teaching biology are my hobbies and passion, it is a continuous journey, and I welcome you all to join with me Archives
July 2022
Categories
All



|
 RSS Feed
RSS Feed
